

Mucoviscidose : vers un rétablissement des fonctions respiratoires ?
Les résultats d’une toute récente étude menée par des chercheurs français ouvrent une piste thérapeutique dans la mucoviscidose. Avec à terme, l’espoir de permettre aux malades de recouvrer leur fonction respiratoire.
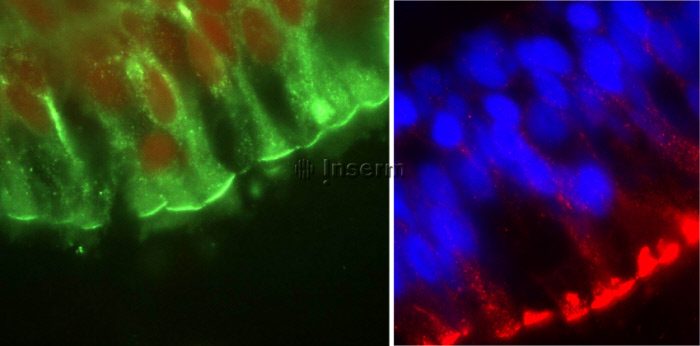
Maladie d’origine génétique, la mucoviscidose touche en moyenne un nouveau-né sur 4 500. Les progrès de la recherche ont permis d’identifier le gène responsable. Situé sur le chromosome 7, il code pour la protéine CFTR (pour Cystic Fibrosis transmembrane Conductance Regulator). Cette protéine fonctionne comme un canal membranaire, permettant l’échange d’ions chlorure entre l’intérieur et l’extérieur des cellules.
Lorsque une mutation se produit sur le gène codant, la protéine produite devient alors déficiente. Résultat : le canal ne fonctionne plus. Au niveau de la muqueuse pulmonaire, un tel dysfonctionnement induit des infections et inflammations chroniques. Ces symptômes traduisent la mucoviscidose. À terme, ils causent la destruction de la muqueuse pulmonaire du malade, entraînant le décès. À ce jour, l’espérance de vie est d’environ 40 ans.
Rétablir la circulation du chlorure
La difficulté première dans l’obtention d’un traitement repose sur le fait que plus de 2 000 mutations du gène CFTR ont été identifiées. Difficile, pour ne pas dire impossible, de trouver une approche thérapeutique globale efficace face à autant de modifications différentes possibles. Sauf à trouver un moyen de se passer de la protéine CFTR. C’est exactement la stratégie poursuivie par une équipe de chercheurs du Centre de recherche Saint-Antoine (Inserm 938 et Université Pierre et Marie Curie).
Ils ont mis en évidence un mécanisme indépendant de CFTR, permettant de rétablir le fonctionnement d’un canal chlorure, dénommé Anoctamin-1 (ANO1). Ce canal est une cible thérapeutique de choix car il se situe notamment dans la muqueuse pulmonaire. Afin d’activer ANO1, les scientifiques ont synthétisé une molécule capable de bloquer l’inhibition (la fermeture) du canal afin qu’il laisse passer les ions.
« Cibler l’ensemble des patients atteints de mucoviscidose. »
Il faut imaginer le canal chlorure comme une porte munie d’une serrure. Quand la clé, représentée par l’inhibiteur, rentre dans la serrure, la porte se ferme. Or, la molécule mise au point par les chercheurs français empêche la clé de rentrer dans la serrure. Du coup, la porte (donc le canal ) peut continuer à s’ouvrir pour laisser passer les ions.
Comme l’explique Olivier Tabary, un des auteurs de l’étude parue dans la revue Nature Communications, « par cette technique, nous avons pu rétablir dans des cultures de cellules de patients atteints de mucoviscidose, les efflux chlorures, la réparation tissulaire ainsi que la clairance muco-ciliaire ». Des paramètres cruciaux dans l’évolution de la maladie. « Une telle stratégie, poursuit le chercheur, permettrait de cibler à terme l’ensemble des patients quelle que soit la mutation et de corriger des paramètres majeurs dans le développement de leur physiopathologie. » O. Clot-Faybesse

Vos avantages :
- Magazine téléchargeable en ligne tous les 2 mois (format PDF)
- Accès à tous les articles du site internet
- Guides pratiques à télécharger
- 2 ans d’archives consultables en ligne
A lire également
![[En salles] La Machine à écrire et autres sources de tracas : petits objets réparés, grands soucis](https://www.faire-face.fr/wp-content/uploads/fly-images/la-machine-a-ecrire-slider-383x168-cc.png)
[En salles] La Machine à écrire et autres sources de tracas : petits objets réparés, grands soucis écartés

Polyarthrite rhumatoïde : l’abatacept, une molécule pour prévenir la maladie
